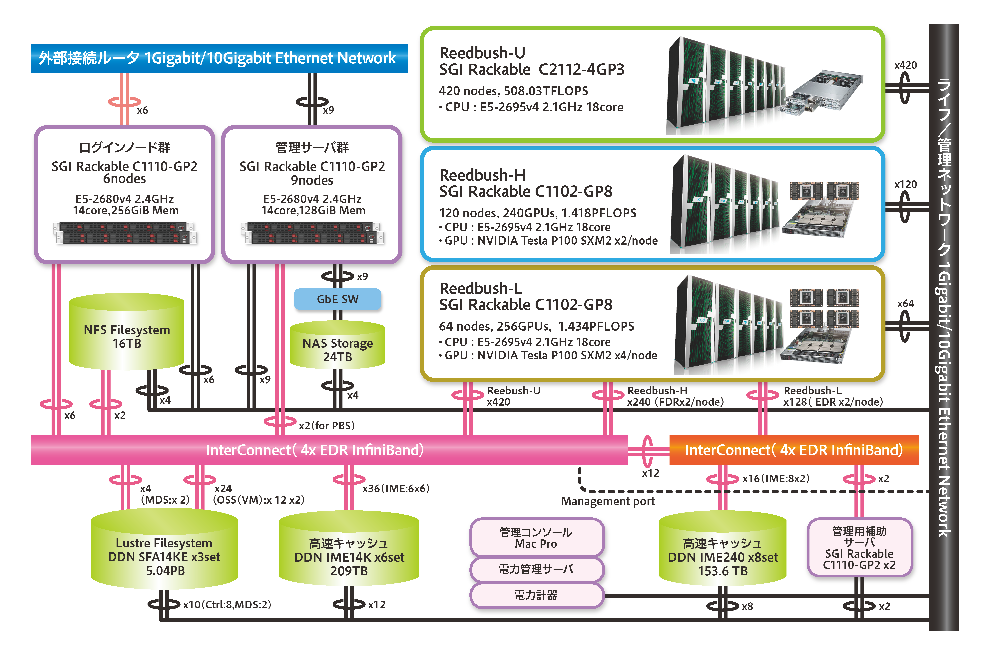
今回から、Reedbushスーパーコンピュータシステムを利用するので、様々な解析ツールのインストールを行い、解析環境を整えます。
備忘録
# sshでログインノードにアクセスする。
$ ssh j29XXX@reedbush.cc.u-tokyo.ac.jp
# qsubコマンドで処理を実行するための計算機(計算ノード)にアクセスする。
[j29XXX@reedbush-h1 ~]$ qsub ...
# ログインノードのみアクセス可能な記憶領域。ssh等のログインに必要な設定ファイルなど、容量の比較的小さいファイルのみ置いてある。
[j29XXX@rreedbush-h1 ~]$ cd /home/gj29/j29XXX
# 学科全体で利用可能な高速アクセス領域。計算ノードのホームディレクトリ、ログインノードからアクセス可能。
[j29XXX@rreedbush-h1 ~]$ cd /lustre/gj29/j29XXXOSの確認
ディストリビューションの確認
[j29XXX@reedbush-u4 j29XXX]$ cat /etc/redhat-release
Red Hat Enterprise Linux Server release 7.4 (Maipo)Red Hat は CentOS系(正確には、CentOSはRed Hat)なので、今回は CentOS系統のものをダウンロードします。
ちなみに、以下のコマンドを打てば、LinuxOSのどのディストリビューションを使っているか調べることができます。
$ cat /etc/*release # Ubuntu: /etc/lsb-release # Fedora: /etc/fedora-release
$ cat /etc/*version # Debin: /etc/debian_version # CentOS / RedHat Enterprise: /etc/redhat-releaseその他カーネル情報の確認
[j29XXX@reedbush-u4 j29XXX]$ uname -a
Linux reedbush-u4 3.10.0-693.11.6.el7.x86_64 #1 SMP Thu Dec 28 14:23:39 EST 2017 x86_64 x86_64 x86_64 GNU/Linux64-bit バージョンの x86 instruction set であることがわかります。
各種ツールのダウンロード
今回は、以下のツールをダウンロードします。
| ツール名 | 説明 | ウェブサイト |
|---|---|---|
| SRA Toolkit | sra形式の実験データの取得、fastq形式の塩基配列データへの変換など | https://www.ncbi.nlm.nih.gov/sra/docs/toolkitsoft/ |
| FASTQC | 配列データの品質チェック | https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ |
| HISAT2 | スプライシングを考慮した高速な配列マッピング | https://ccb.jhu.edu/software/hisat2/index.shtml |
| Samtools | マッピング結果ファイルの各種操作 | http://www.htslib.org/download/ |
| IGV | マッピング結果をグラフィカルに表示するビューワ(自分のPCにインストールして使用) | http://software.broadinstitute.org/software/igv/download |
| Subread | リードの高速カウントを行う featureCounts などを含むパッケージ | http://subread.sourceforge.net/ |
SRA Toolkit
sra形式の実験データの取得、fastq形式の塩基配列データへの変換などを行うツール。
[j29XXX@reedbush-u4 j29XXX]$ wget https://ftp-trace.ncbi.nlm.nih.gov/sra/sdk/current/sratoolkit.current-centos_linux64.tar.gz
[j29XXX@reedbush-u4 j29XXX]$ tar -zxvf sratoolkit.current-centos_linux64.tar.gz
[j29XXX@reedbush-u4 j29XXX]$ ls
sratoolkit.2.9.6-1-centos_linux64
sratoolkit.current-centos_linux64.tar.gz
[j29XXX@reedbush-u4 j29XXX]$ cd sratoolkit.2.9.6-1-centos_linux64/bin
# 以下のコマンドで動けば成功です。
[j29XXX@reedbush-u4 bin]$ ./fasterq-dump --version # fastqファイルを取得する時に使うコマンドです。FASTQC
配列データの品質チェックを行うツール。
[j29XXX@reedbush-u4 j29XXX]$ wget https://www.bioinformatics.babraham.ac.uk/projects/fastqc/fastqc_v0.11.8.zip
[j29XXX@reedbush-u4 j29XXX]$ unzip fastqc_v0.11.8.zip
[j29XXX@reedbush-u4 j29XXX]$ ls
FastQC
fastqc_v0.11.8.zip
# 実行ファイルに変更する。
[j29XXX@reedbush-u4 j29XXX]$ cd FastQC
[j29XXX@reedbush-u4 FastQC]$ chmod 755 fastqc
# 以下のコマンドで動けば成功です。
[j29XXX@reedbush-u4 FastQC]$ ./fastqc --version
# もし動かなければ、Javaがインストールされていないのかもしれません。以下コマンドで確認できます。
# (FastQC is a java application. In order to run it needs your system to have a suitable Java Runtime Environment (JRE) installed.)
[j29XXX@reedbush-u4 j29XXX]$ java -versionHISAT2
スプライシングを考慮した高速な配列マッピングをするツール。
[j29XXX@reedbush-u4 j29XXX]$ wget ftp://ftp.ccb.jhu.edu/pub/infphilo/hisat2/downloads/hisat2-2.1.0-Linux_x86_64.zip
[j29XXX@reedbush-u4 j29XXX]$ unzip hisat2-2.1.0-Linux_x86_64.zip
[j29XXX@reedbush-u4 j29XXX]$ ls
hisat2-2.1.0
hisat2-2.1.0-Linux_x86_64.zip
[j29XXX@reedbush-u4 j29XXX]$ cd hisat2-2.1.0
# 以下のコマンドで動けば成功です。
[j29XXX@reedbush-u4 hisat2-2.1.0]$ ./hista2 --versionSamtools
マッピング結果ファイルの各種操作を行うツール。
[j29XXX@reedbush-u4 j29XXX]$ wget https://github.com/samtools/samtools/releases/download/1.9/samtools-1.9.tar.bz2
[j29XXX@reedbush-u4 j29XXX]$ tar -jxvf samtools-1.9.tar.bz2
[j29XXX@reedbush-u4 j29XXX]$ ls
samtools-1.9
samtools-1.9.tar.bz2
[j29XXX@reedbush-u4 j29XXX]$ cd samtools-1.9
[j29XXX@reedbush-u4 samtools-1.9]$ ./configure --prefix=/lustre/gjXX/j29XXX
[j29XXX@reedbush-u4 samtools-1.9]$ make
[j29XXX@reedbush-u4 samtools-1.9]$ make install
# 以下のコマンドで動けば成功です。
[j29XXX@reedbush-u4 samtools-1.9]$ ./samtools --versionIGV
マッピング結果をグラフィカルに表示するビューワ。 ※このツールはGUIツールなので、ここでは割愛します。
Subread
リードの高速カウントを行う featureCounts などを含むパッケージ
[j29XXX@reedbush-u4 j29XXX]$ wget --content-disposition https://sourceforge.net/projects/subread/files/subread-2.0.0/subread-2.0.0-Linux-x86_64.tar.gz/download
[j29XXX@reedbush-u4 j29XXX]$ tar -zxvf subread-2.0.0-Linux-x86_64.tar.gz
[j29XXX@reedbush-u4 j29XXX]$ ls
subread-2.0.0-Linux-x86_64
subread-2.0.0-Linux-x86_64.tar.gz
[j29XXX@reedbush-u4 j29XXX]$ cd subread-2.0.0-Linux-x86_64/bin
# 以下のコマンドで動けば成功です。
[j29XXX@reedbush-u4 bin]$ ./featureCountsパスを通す
いちいちパスを通すのが面倒なので、.bashrc の環境変数にパスを書き込みます。
[j29XXX@reedbush-u4 j29XXX]$ vi .bashrc # エディタはなんでも良いので以下を書き込む
""".bashrc
PATH=$PATH:/lustre/gjXX/j29XXX/sratoolkit.2.9.6-1-centos_linux64/bin
PATH=$PATH:/lustre/gjXX/j29XXX/FastQC
PATH=$PATH:/lustre/gjXX/j29XXX/hisat2-2.1.0
PATH=$PATH:/lustre/gjXX/j29XXX/samtools-1.9
PATH=$PATH:/lustre/gjXX/j29XXX/subread-2.0.0-Linux-x86_64/bin
"""
[j29XXX@reedbush-u4 j29XXX]$ source .bashrc # shellの更新もしくは、以下のコマンドを打ちます。
[j29XXX@reedbush-u4 j29XXX]$ export PATH=$PATH:/lustre/gjXX/j29XXX/sratoolkit.2.9.6-1-centos_linux64/bin
[j29XXX@reedbush-u4 j29XXX]$ export PATH=$PATH:/lustre/gjXX/j29XXX/FastQC
[j29XXX@reedbush-u4 j29XXX]$ export PATH=$PATH:/lustre/gjXX/j29XXX/hisat2-2.1.0
[j29XXX@reedbush-u4 j29XXX]$ export PATH=$PATH:/lustre/gjXX/j29XXX/samtools-1.9
[j29XXX@reedbush-u4 j29XXX]$ export PATH=$PATH:/lustre/gjXX/j29XXX/subread-2.0.0-Linux-x86_64/binこれにより、どこからでも各種解析ツールを呼び出すことができました。試しに samtools を呼び出してみます。
# 以下のコマンドで動けば成功です。
[j29XXX@reedbush-u4 j29XXX]$ samtools --version