teilab.normalizations module¶
This submodule contains various functions and classes that are useful for normalization.
Instructions¶
Differential gene expression can be an outcome of true biological variability or experimental artifacts. Normalization techniques have been used to minimize the effect of experimental artifacts on differential gene expression analysis.
Robust Multichip Analysis (RMA)¶
In microarray analysis, many algorithms have been proposed, but the most widely used one (de facto standard) is Robust Multichip Analysis (RMA) , where the signal value of each spot ( RawData ) is processed and normalized according to the following flow. ( 1) Background Subtraction, 2) Normalization Between Samples and 3) Summarization )
![digraph RAMPreprocessingGraph {
graph [
charset = "UTF-8";
label = "Preprocessing (RMA)",
labelloc = "t",
labeljust = "c",
bgcolor = "#1f441e",
fontcolor = "white",
fontsize = 18,
style = "filled",
rankdir = TB,
margin = 0.2,
ranksep = 1.0,
nodesep = 0.9,
layout = dot,
compound = true,
];
node [
style = "solid,filled",
fontsize = 16,
fontcolor = 6,
fontname = "monospace",
color = "#cee6b4",
fillcolor = "#9ecca4",
fixedsize = false,
margin = "0.2,0.1",
];
RawData_row1_col1 [shape=doublecircle margin="0" fillcolor="#29bb89" fontcolor="#be0000" color="#29bb89" label="RawData\n(row1,col1)"];
RawData_row1_col2 [shape=doublecircle margin="0" fillcolor="#29bb89" fontcolor="#be0000" color="#29bb89" label="RawData\n(row1,col2)"];
RawData_rowN_colM [shape=doublecircle margin="0" fillcolor="#29bb89" fontcolor="#be0000" color="#29bb89" label="RawData\n(rowN,colM)"];
Ave_row1_col1 [shape=circle margin="0" fontcolor="#233e8b" fillcolor="#b6c9f0" color="#233e8b" label="ave"];
Ave_row1_col2 [shape=circle margin="0" fontcolor="#233e8b" fillcolor="#b6c9f0" color="#233e8b" label="ave"];
Ave_rowN_colM [shape=circle margin="0" fontcolor="#233e8b" fillcolor="#b6c9f0" color="#233e8b" label="ave"];
gMeanSignal_row1_col1 [
shape = none
margin = 0
label = <
<table width="100" border="0" cellspacing="10" bgcolor="white">
<tr><td port="varname" align="left" border="1" color="#e1e4e5" bgcolor="white" width="110" fixedsize="true"><font point-size="16" face="monaco" color="#e74c3c">gMeanSignal</font></td></tr>
<tr><td port="location"><font face="Courier">row1,col1</font></td></tr>
</table>
>
]
gMeanSignal_row1_col2 [
shape = none
margin = 0
label = <
<table width="100" border="0" cellspacing="10" bgcolor="white">
<tr><td port="varname" align="left" border="1" color="#e1e4e5" bgcolor="white" width="110" fixedsize="true"><font point-size="16" face="monaco" color="#e74c3c">gMeanSignal</font></td></tr>
<tr><td port="location"><font face="Courier">row1,col2</font></td></tr>
</table>
>
]
gMeanSignal_rowN_colM [
shape = none
margin = 0
label = <
<table width="100" border="0" cellspacing="10" bgcolor="white">
<tr><td port="varname" align="left" border="1" color="#e1e4e5" bgcolor="white" width="110" fixedsize="true"><font point-size="16" face="monaco" color="#e74c3c">gMeanSignal</font></td></tr>
<tr><td port="location"><font face="Courier">rowN,colM</font></td></tr>
</table>
>
]
gBGUsed_row1_col1 [
shape = none
margin = 0
label = <
<table width="100" border="0" cellspacing="10" bgcolor="white">
<tr><td port="varname" align="left" border="1" color="#e1e4e5" bgcolor="white" width="80" fixedsize="true"><font point-size="16" face="monaco" color="#e74c3c">gBGUsed</font></td></tr>
<tr><td port="location"><font face="Courier">row1,col1</font></td></tr>
</table>
>
]
gBGUsed_row1_col2 [
shape = none
margin = 0
label = <
<table width="100" border="0" cellspacing="10" bgcolor="white">
<tr><td port="varname" align="left" border="1" color="#e1e4e5" bgcolor="white" width="80" fixedsize="true"><font point-size="16" face="monaco" color="#e74c3c">gBGUsed</font></td></tr>
<tr><td port="location"><font face="Courier">row1,col2</font></td></tr>
</table>
>
]
gBGUsed_rowN_colM [
shape = none
margin = 0
label = <
<table width="100" border="0" cellspacing="10" bgcolor="white">
<tr><td port="varname" align="left" border="1" color="#e1e4e5" bgcolor="white" width="80" fixedsize="true"><font point-size="16" face="monaco" color="#e74c3c">gBGUsed</font></td></tr>
<tr><td port="location"><font face="Courier">rowN,colM</font></td></tr>
</table>
>
]
gBGSubSignal [shape=box fontname="monaco" fontcolor="#e74c3c" fillcolor="white" color="#e1e4e5"];
rBGSubSignal [shape=box fontname="monaco" fontcolor="#e74c3c" fillcolor="white" color="#e1e4e5"];
gDyeNormSignal [shape=box fontname="monaco" fontcolor="#e74c3c" fillcolor="white" color="#e1e4e5"];
rDyeNormSignal [shape=box fontname="monaco" fontcolor="#e74c3c" fillcolor="white" color="#e1e4e5"];
Minus [shape=circle margin="0" fontcolor="#233e8b" fillcolor="#b6c9f0" color="#233e8b" label="-"];
BackgroundSubtraction [shape=diamond fontname="fantasy" label="Background Subtraction\n(+ Detrended fluctuation Analysis)"];
DyeNormalization [shape=diamond fontname="fantasy" margin="0.05" label="(Dye Normalization)"];
SurrogateVariableAnalysis [shape=diamond fontname="fantasy" margin="0.05" label="Surrogate Variable Analysis"];
QuantileNormalization [shape=diamond fontname="fantasy" margin="0.05" label="Quantile Normalization"];
Summarization [shape=diamond fontname="fantasy" margin="0.05" label="Summarization"];
remaining_processing_row1_col2 [shape=Mdiamond fontname="fantasy" margin="0.05" label="Remaining Processing"]
remaining_processing_rowN_colM [shape=Mdiamond fontname="fantasy" margin="0.05" label="Remaining Processing"]
gProcessedSignal_vimentin1 [
shape = none
margin = 0
label = <
<table width="100" border="0" cellspacing="10" bgcolor="#fff5b7">
<tr><td port="varname" align="left" border="1" color="#e1e4e5" bgcolor="white" width="160" fixedsize="true"><font point-size="16" face="monaco" color="#e74c3c">gProcessedSignal</font></td></tr>
<tr><td port="gene"><font face="Courier">vimentin1</font></td></tr>
</table>
>
]
gProcessedSignal_insulin1 [
shape=none
margin = 0
label = <
<table width="100" border="0" cellspacing="10" bgcolor="#fff5b7">
<tr><td port="varname" align="left" border="1" color="#e1e4e5" bgcolor="white" width="160" fixedsize="true"><font point-size="16" face="monaco" color="#e74c3c">gProcessedSignal</font></td></tr>
<tr><td port="gene"><font face="Courier">insulin1</font></td></tr>
</table>
>
]
gProcessedSignal_vimentin3 [
shape = none
margin = 0
label = <
<table width="100" border="0" cellspacing="10" bgcolor="#fff5b7">
<tr><td port="varname" align="left" border="1" color="#e1e4e5" bgcolor="white" width="160" fixedsize="true"><font point-size="16" face="monaco" color="#e74c3c">gProcessedSignal</font></td></tr>
<tr><td port="gene"><font face="Courier">vimentin3</font></td></tr>
</table>
>
]
gProcessedSignals_sampleA [
shape = none
margin = 0
label = <
<table width="100" border="0" cellspacing="10" bgcolor="#ffc93c">
<tr>
<td rowspan="2" port="varname" align="left" border="1" color="#e1e4e5" bgcolor="white" width="170" fixedsize="true"><font point-size="16" face="monaco" color="#e74c3c">gProcessedSignals</font></td>
<td port="vim1"><font face="Courier">vimentin1</font></td>
</tr>
<tr><td port="ins1"><font face="Courier">insulin1</font></td></tr>
<tr bgcolor="red">
<td rowspan="2" port="varname" align="center" border="0" width="170" fixedsize="true"><font point-size="26" color="#1f441e">SAMPLE_A</font></td>
<td port="ins1"><font face="Courier">:</font></td>
</tr>
<tr><td port="vim3"><font face="Courier">vimentin3</font></td></tr>
</table>
>
]
gProcessedSignals_sampleB [
shape = none
margin = 0
label = <
<table width="100" border="0" cellspacing="10" bgcolor="#ffc93c">
<tr>
<td rowspan="2" port="varname" align="left" border="1" color="#e1e4e5" bgcolor="white" width="170" fixedsize="true"><font point-size="16" face="monaco" color="#e74c3c">gProcessedSignals</font></td>
<td port="vim1"><font face="Courier">vimentin1</font></td>
</tr>
<tr><td port="ins1"><font face="Courier">insulin1</font></td></tr>
<tr bgcolor="red">
<td rowspan="2" port="varname" align="center" border="0" width="170" fixedsize="true"><font point-size="26" color="#1f441e">SAMPLE_B</font></td>
<td port="ins1"><font face="Courier">:</font></td>
</tr>
<tr><td port="vim3"><font face="Courier">vimentin3</font></td></tr>
</table>
>
]
gProcessedSignal_vimentin1_normed [
shape = none
margin = 0
label = <
<table width="100" border="0" cellspacing="10" bgcolor="#fff5b7">
<tr><td port="varname" align="left" border="1" color="#e1e4e5" bgcolor="white" width="230" fixedsize="true"><font point-size="16" face="monaco" color="#e74c3c">gProcessedSignal_normed</font></td></tr>
<tr><td port="gene"><font face="Courier">vimentin1</font></td></tr>
</table>
>
]
gProcessedSignal_insulin1_normed [
shape=none
margin = 0
label = <
<table width="100" border="0" cellspacing="10" bgcolor="#fff5b7">
<tr><td port="varname" align="left" border="1" color="#e1e4e5" bgcolor="white" width="230" fixedsize="true"><font point-size="16" face="monaco" color="#e74c3c">gProcessedSignal_normed</font></td></tr>
<tr><td port="gene"><font face="Courier">insulin1</font></td></tr>
</table>
>
]
gProcessedSignal_vimentin3_normed [
shape = none
margin = 0
label = <
<table width="100" border="0" cellspacing="10" bgcolor="#fff5b7">
<tr><td port="varname" align="left" border="1" color="#e1e4e5" bgcolor="white" width="230" fixedsize="true"><font point-size="16" face="monaco" color="#e74c3c">gProcessedSignal_normed</font></td></tr>
<tr><td port="gene"><font face="Courier">vimentin3</font></td></tr>
</table>
>
]
gProcessedSignal_vimentin_normed [
shape = none
margin = 0
label = <
<table width="100" border="0" cellspacing="10" bgcolor="#fff5b7">
<tr><td port="varname" align="left" border="1" color="#e1e4e5" bgcolor="white" width="230" fixedsize="true"><font point-size="16" face="monaco" color="#e74c3c">gProcessedSignal_normed</font></td></tr>
<tr><td port="gene"><font face="Courier">vimentin</font></td></tr>
</table>
>
]
gProcessedSignals_sampleB_normed [
shape = none
margin = 0
label = <
<table width="100" border="0" cellspacing="10" bgcolor="#ffc93c">
<tr>
<td rowspan="2" port="varname" align="left" border="1" color="#e1e4e5" bgcolor="white" width="240" fixedsize="true"><font point-size="16" face="monaco" color="#e74c3c">gProcessedSignals_normed</font></td>
<td port="vim1"><font face="Courier">vimentin1_normed</font></td>
</tr>
<tr><td port="ins1"><font face="Courier">insulin1_normed</font></td></tr>
<tr bgcolor="red">
<td rowspan="2" port="varname" align="center" border="0" width="170" fixedsize="true"><font point-size="26" color="#1f441e">SAMPLE_B</font></td>
<td port="ins1"><font face="Courier">:</font></td>
</tr>
<tr><td port="vim3"><font face="Courier">vimentin3_normed</font></td></tr>
</table>
>
]
gProcessedSignals_sampleA_normed [
shape = none
margin = 0
label = <
<table width="100" border="0" cellspacing="10" bgcolor="#ffc93c">
<tr>
<td rowspan="2" port="varname" align="left" border="1" color="#e1e4e5" bgcolor="white" width="240" fixedsize="true"><font point-size="16" face="monaco" color="#e74c3c">gProcessedSignals_normed</font></td>
<td port="vim1"><font face="Courier">vimentin1_normed</font></td>
</tr>
<tr><td port="ins1"><font face="Courier">insulin1_normed</font></td></tr>
<tr bgcolor="red">
<td rowspan="2" port="varname" align="center" border="0" width="170" fixedsize="true"><font point-size="26" color="#1f441e">SAMPLE_A</font></td>
<td port="ins1"><font face="Courier">:</font></td>
</tr>
<tr><td port="vim3"><font face="Courier">vimentin3_normed</font></td></tr>
</table>
>
]
edge [
style = solid,
fontsize = 14,
fontcolor = white,
fontname = "Migu 1M",
color = white,
labelfloat = true,
labeldistance = 2.5,
labelangle = 70
];
RawData_row1_col1 -> Ave_row1_col1 -> gMeanSignal_row1_col1;
RawData_row1_col2 -> Ave_row1_col2 -> gMeanSignal_row1_col2;
RawData_rowN_colM -> Ave_rowN_colM -> gMeanSignal_rowN_colM;
subgraph cluster_1_row1_col1 {
label = "Background Subtraction";
labelloc = "t";
labeljust = "c";
fillcolor = "#89898989";
fontcolor = "#ffd56b";
margin = 20;
gMeanSignal_row1_col1:varname -> BackgroundSubtraction
BackgroundSubtraction -> gBGUsed_row1_col1;
gMeanSignal_row1_col1 -> Minus;
gBGUsed_row1_col1 -> Minus;
Minus -> gBGSubSignal;
gBGSubSignal -> DyeNormalization;
rBGSubSignal -> DyeNormalization;
DyeNormalization -> gDyeNormSignal;
DyeNormalization -> rDyeNormSignal;
gDyeNormSignal -> SurrogateVariableAnalysis;
SurrogateVariableAnalysis -> gProcessedSignal_vimentin1;
};
subgraph cluster_1_row1_col2 {
label = "Background Subtraction";
labelloc = "b";
labeljust = "l";
fillcolor = "#89898989";
fontcolor = "#ffd56b";
margin = 20;
gMeanSignal_row1_col2 -> BackgroundSubtraction -> gBGUsed_row1_col2 -> remaining_processing_row1_col2;
gMeanSignal_row1_col2 -> remaining_processing_row1_col2;
remaining_processing_row1_col2 -> gProcessedSignal_insulin1;
}
subgraph cluster_1_rowN_colM {
label = "Background Subtraction";
labelloc = "b";
labeljust = "r";
fillcolor = "#89898989";
fontcolor = "#ffd56b";
margin = 20;
gMeanSignal_rowN_colM -> BackgroundSubtraction -> gBGUsed_rowN_colM -> remaining_processing_rowN_colM;
gMeanSignal_rowN_colM -> remaining_processing_rowN_colM
remaining_processing_rowN_colM -> gProcessedSignal_vimentin3
}
gProcessedSignal_vimentin1 -> gProcessedSignals_sampleA:vim1
gProcessedSignal_vimentin3 -> gProcessedSignals_sampleA:vim3
gProcessedSignal_insulin1 -> gProcessedSignals_sampleA:ins1
subgraph cluster_2 {
label = "Normalization Between Samples";
labelloc = "t";
labeljust = "l";
fillcolor = "#89898989";
fontcolor = "#ffd56b";
margin = 20;
gProcessedSignals_sampleA -> QuantileNormalization;
gProcessedSignals_sampleB -> QuantileNormalization;
QuantileNormalization -> gProcessedSignals_sampleB_normed;
QuantileNormalization -> gProcessedSignals_sampleA_normed;
};
gProcessedSignals_sampleA_normed:vim1 -> gProcessedSignal_vimentin1_normed;
gProcessedSignals_sampleA_normed:ins1 -> gProcessedSignal_insulin1_normed;
gProcessedSignals_sampleA_normed:vim3 -> gProcessedSignal_vimentin3_normed;
subgraph cluster_3 {
label = "Summarization";
labelloc = "t";
labeljust = "l";
fillcolor = "#89898989";
fontcolor = "#ffd56b";
margin = 20;
gProcessedSignal_vimentin1_normed -> Summarization;
gProcessedSignal_vimentin3_normed -> Summarization;
Summarization -> gProcessedSignal_vimentin_normed;
};
}](_images/graphviz-7d9910d4a36548f0df6bfc64bf2016bd419a5dbf.png)
1. Background Subtraction¶
In Background Subtraction, we assume that the observed signal intensity is a combnation of the actual signal intensity and the background signal intensity (derived from Non-specific Hybridization), and aim to eliminate the influence of the latter one.
In the general method, we make assumptions such as
Actual signal intensity distribution is an exponential distribution.
Background signal intensity distribution is a normal distribution.
Then, by optimizing the parameters to best represent the phenomenon, the background intensity distribution is calculated and subtracted.
2. Normalization Between Samples¶
Here, we perform normalization “between” samples. What can be said from the results of a “single sample” microarray experiment are very limited, and we should compare with other (treatment sample, control group, etc.) experimental results. Howevet, bias due to experimental operation and equipment characteristics is inevitable, and if you just compare them as they are, you will misinterpret them.
For example, if you want to characterize the changes in global gene expression in the livers of H1/siRNAinsulin-CMV/hIDE transgenic (Tg) mice in response to the reduced bioavailability of insulin 1, and the expression level of each RNA in Tg mice was generally lower than that of non-Tg mice, you may mistakenly conclude that almost all of the RNAs were down-regulated respectively by reduced bioavailability of insulin.
Therefore, it is necessary to reduce the influence of the bias. There are numerous proposals for normalizing unbalanced data between samples ( This review paper summarizes 23 normalization methods developed for unbalanced transcriptome data), but each method makes some assumptions in data, so it is important to choose the correct normalization method for each experimental results.
We will introduce two majour methods.
Note
1. Percentile¶
This method is a constant adjustoment and the most straightforward.
Calculate the x%tile for each distribution.
Average them.
Divide each distribution by its x%tile and multiply by averaged value.
Example |
|---|
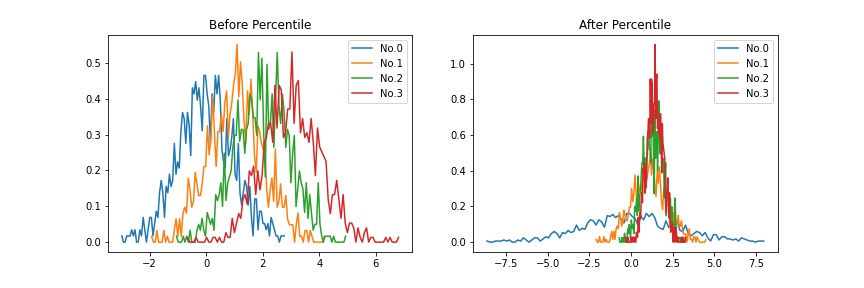
|
Defined as percentile in this package.
2. Quantile¶
Quantile Normalization is a technique for making all distributions identical in statistical properties. It was introduced as “quantile standardization” (in Analysis of Data from Viral DNA Microchips ) and then renamed as “quantile normalization” (in A comparison of normalization methods for high density oligonucleotide array data based on variance and bias )
To quantile normalize the all distributions,
Sort each distribution.
Average the data in the same rank.
Replace the value of each rank with the averaged value.
Warning
This method can be used when the assumption that “the intensity distribution of gene expression in each sample is almost the same” holds.
Example |
|---|
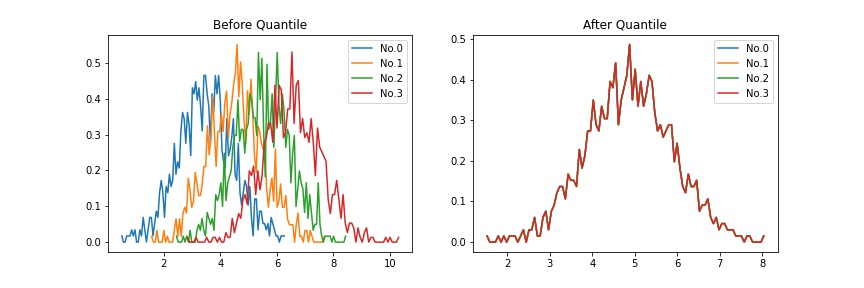
|
Defined as quantile in this package.
Python Objects¶
- teilab.normalizations.percentile(data: nptyping.types._ndarray.NDArray[Any, Any, nptyping.types._object.Object], percent: numbers.Number = 75) nptyping.types._ndarray.NDArray[Any, Any, nptyping.types._object.Object][source]¶
Perform Percentile Normalization.
- Parameters
data (NDArray[(Any,Any),Number]) – Input data. Shape = (
n_samples,n_features)percent (Number, optional) – Which percentile value to normalize. Defaults to
75.
- Returns
percentiled data. Shape = (
n_samples,n_features)- Return type
NDArray[(Any,Any),Number]
- Raises
ValueError – When
percenttiles contain negative values.
Examples
>>> import matplotlib.pyplot as plt >>> from teilab.normalizations import percentile >>> from teilab.plot.matplotlib import densityplot >>> n_samples, n_features = (4, 1000) >>> data = np.random.RandomState(0).normal(loc=np.expand_dims(np.arange(n_samples), axis=1), size=(n_samples,n_features)) >>> data_percentiled = percentile(data=data, percent=75) >>> fig, axes = plt.subplots(ncols=2, nrows=1, figsize=(12,4)) >>> ax = densityplot(data=data, title="Before Percentile", ax=axes[0]) >>> ax = densityplot(data=data_percentiled, title="After Percentile", ax=axes[1])
Results
- teilab.normalizations.quantile(data: nptyping.types._ndarray.NDArray[Any, Any, nptyping.types._object.Object]) nptyping.types._ndarray.NDArray[Any, Any, nptyping.types._object.Object][source]¶
Perform Quantile Normalization.
- Parameters
data (NDArray[(Any,Any),Number]) – Input data. Shape = (
n_samples,n_features)- Returns
percentiled data. Shape = (
n_samples,n_features)- Return type
NDArray[(Any,Any),Number]
- Raises
ValueError – When
datacontains negative values.
Examples
>>> import matplotlib.pyplot as plt >>> from teilab.normalizations import quantile >>> from teilab.plot.matplotlib import densityplot >>> n_samples, n_features = (4, 1000) >>> data = np.random.RandomState(0).normal(loc=np.expand_dims(np.arange(n_samples), axis=1), size=(n_samples,n_features), ) + 3.5 >>> data_quantiled = quantile(data=data) >>> fig, axes = plt.subplots(ncols=2, nrows=1, figsize=(12,4)) >>> ax = densityplot(data=data, title="Before Quantile", ax=axes[0]) >>> ax = densityplot(data=data_quantiled, title="After Quantile", ax=axes[1])
Results
- teilab.normalizations.median_polish(data: nptyping.types._ndarray.NDArray[Any, Any, nptyping.types._object.Object], labels: nptyping.types._ndarray.NDArray[Any, Any], rtol: float = 1e-05, atol: float = 1e-08) nptyping.types._ndarray.NDArray[Any, Any, nptyping.types._object.Object][source]¶
Median Polish
- Parameters
data (NDArray[(Any,Any),Number]) – Input data. Shape = (
n_samples,n_features)labels (NDArray[(Any),Any]) – Label (ex.
GeneName, orSystematicName)rtol (float) – The relative tolerance parameter. Defaults to
1e-05atol (float) – The absolute tolerance parameter. Defaults to
1e-08
- Raises
TypeError – When
data.shape[1]is not the same aslen(labels)- Returns
Median Polished Data.
- Return type
NDArray[(Any,Any),Number]
Examples
>>> from teilab.normalizations import median_polish >>> data = np.asarray([ ... [16.1, 14.6, 19.6, 13.6, 13.6, 13.6], ... [ 9.0, 18.4, 6.7, 11.1, 6.7, 9.0], ... [22.4, 13.6, 22.4, 6.7, 9.0, 3.0], >>> ], dtype=float).T >>> n_samples, n_features = data.shape >>> # This means that "All spots (features) are for the same gene." >>> labels = np.zeros(shape=n_features, dtype=np.uint8) >>> median_polish(data, labels=labels) array([[16.1 , 11.25, 13.3 ], [16.4 , 11.55, 13.6 ], [19.6 , 14.75, 16.8 ], [13.6 , 8.75, 10.8 ], [11.8 , 6.95, 9. ], [13.6 , 8.75, 10.8 ]])
Todo
Speed-UP using
joblib,multiprocessing,Cython, etc.
- teilab.normalizations.median_polish_group_wise(data: pandas.core.generic.NDFrame, rtol: float = 1e-05, atol: float = 1e-08) pandas.core.generic.NDFrame[source]¶
Apply Median polish group-wise.
- Parameters
data (NDFrame) – Input data. Shape = (
n_samples,n_features)rtol (float, optional) – [description]. Defaults to
1e-05.atol (float, optional) – [description]. Defaults to
1e-08.
- Returns
Median Polished Data.
- Return type
NDFrame
Examples
>>> from teilab.normalizations import median_polish_group_wise >>> data = pd.DataFrame(data=[ ... ["vimentin", 16.1, 14.6, 19.6, 13.6, 13.6, 13.6], ... ["vimentin", 9.0, 18.4, 6.7, 11.1, 6.7, 9.0], ... ["vimentin", 22.4, 13.6, 22.4, 6.7, 9.0, 3.0], >>> ], columns=["GeneName"]+[f"Samle.{i}" for i in range(6)]) >>> data.groupby("GeneName").apply(func=median_polish_group_wise).values array([[16.1 , 16.4 , 19.6 , 13.6 , 11.8 , 13.6 ], [11.25, 11.55, 14.75, 8.75, 6.95, 8.75], [13.3 , 13.6 , 16.8 , 10.8 , 9. , 10.8 ]]) >>> # If you want to see the progress, use tqdm. >>> from tqdm import tqdm >>> tqdm.pandas() >>> data.groupby("GeneName").progress_apply(func=median_polish_group_wise).values